Spatial Biology Education
Spatial synergies: how COMET™ is used to validate spatial transcriptomics data
Posted on:

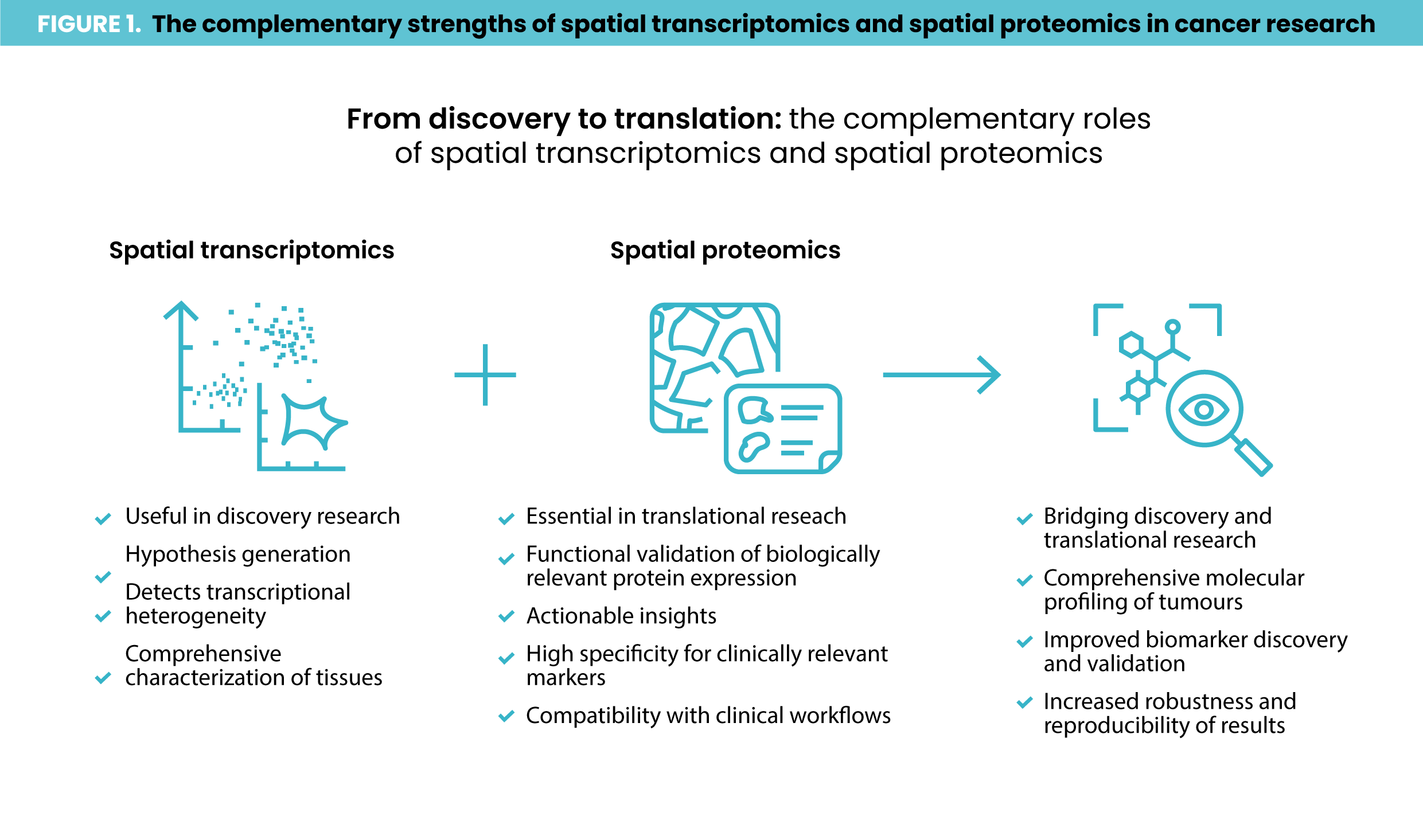
Spatial transcriptomics is a powerful tool for investigating treatment response in oncology and immunotherapy, offering researchers the ability to map RNA activity across tissues with spatial resolution. However, spatial transcriptomics is not yet practical for routine clinical use. This is partly due to its high costs but also because of its complexity, a lack of standardized workflows and limited options for seamless integration with image analysis solutions. So, how can researchers translate spatial transcriptomics findings into clinically actionable assays? One approach is validating spatial transcriptomics discoveries using spatial proteomics, as spatial proteomics aligns closely with widely established clinical assays such as immunohistochemistry (IHC) staining. Proteomic signatures identified through this alignment can be developed into multiplex immunofluorescence (mIF) assays with IHC data serving as a reference standard. Moreover, integrating spatial transcriptomics and spatial proteomics can reveal nuanced biological insights by leveraging the complementary strengths of each technique (Figure 1).
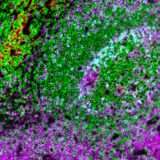
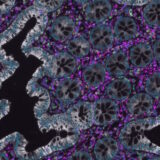
A key factor in enabling this integration is gathering RNA and protein data from the same tissue slide. This approach allows researchers to directly compare gene and protein expression within the same cell at the single-cell level resolution. This concept is closer to reality than many might think. A recent study1 demonstrated the feasibility of combining spatial transcriptomics data from the Xenium platform with spatial proteomics from COMET™ on the same lung cancer tissue section. In the same study the researchers complemented these datasets with histology (H&E staining and annotation) on the cancer tissue sample. The study sought to highlight the potential and practility of conducting spatially-resolved multiomics analysis on the same tissue section without compromising data quality. Combining these analyses enables precise RNA-protein comparisons at the single-cell level and further enhances the clinical applicability of spatial transcriptomics findings. Here’s how the researchers, led by Dr. Joe Yeong‘s lab from the Institute of Molecular and Cell Biology at A*STAR, approached the study.
The power of integrated spatial multiomics
The research team analyzed two lung cancer tissue samples. One underwent spatial transcriptomics, spatial proteomics and H&E staining, while the second underwent spatial proteomics and H&E staining alone (Figure 2). The researchers started by profiling gene expression across the tissue, identifying the expression patterns of 289 genes linked to lung cancer. For spatial transcriptomics, the researchers used used the Xenium in-situ gene expression assay (developed by 10x Genomics), which hybridizes DNA probes to RNA molecules from target genes. The Xenium analyzer platform performed repeated cycles of hybridization, imaging and probe removal to generate optical signatures via gene-specific barcodes. The researchers then examined whether the same tissue slide could undergo spatial proteomics analysis while maintaining data quality. Using the sequential immunofluorescence (seqIF™) assay on the COMET™ platform, they achieved automated cycles of staining, imaging and antibody elution to generate hyperplex immunofluorescence images.